

Free energy is a concept that we have invented to incorporate the entropy change for the surroundings into a change in a state function of the system. Free energy is a way for us to think about the 2nd Law of thermodynamics only from the perspective of the system.
The second law states that for a process to be spontaneous it must increase the entropy of the universe.
\[\Delta S_{\rm univ} = \Delta S_{\rm sys} + \Delta S_{\rm surr} > 0 \]
If we invoke certain conditions, we can rewrite this equation. First we will look at constant pressure, which is a natural condition for chemistry. At constant pressure the heat is simply the change in entropy of the system. Since the change in the entropy of the surrounding is related to the heat, we can now write this in terms of the enthalpy change of the system. We can also look at conditions of constant temperature so the entropy change of the surroundings can simply be related to the enthalpy change of the system.
\[\Delta S_{\rm surr} = {-q \over T} = {- \Delta H_{\rm sys} \over T}\]
We can now rewrite our 2nd Law equation
\[\Delta S_{\rm total} = \Delta S_{\rm sys} + \Delta S_{\rm surr} = \Delta S_{\rm sys} - {\Delta H_{\rm sys} \over T} > 0\]
Now everything is in terms of the system (so we can drop the system subscript). This is, in a way, a small victory. We could be finished here, but our 2nd Law equation is still in entropy terms. We don't like to think in terms of entropy we would rather think in terms of energy. We can get to energy units if we simply multiply through by T.
\[T \Delta S - \Delta H > 0\]
We could again be done here. However, we now have an energy that increases for spontaneous processes. That seems counter intuitive. So we multiply the whole equation by a minus. This yields
\[\Delta H - T \Delta S < 0\]
Introducing a new State Function (variable) - Gibb's Free Energy We now have an energy term for the system which will decrease for a spontaneous process (at constant temperature and pressure). We can define this new state variable to reflect this energy. The new variable is the Gibb's Free Energy, \(G\):
\[ G = H - TS \]
This is the definition of the Gibb's Free Energy which will allow us to predict the spontaneity of a reaction using all system-based variables. This allows us to rewrite the second law based on only a system state function, \(\Delta G\). For a spontaneous process for an isolated system (at constant temperature and pressure) the change in free energy must be negative.
\[{\rm for\;spontaneous\;change: }\hskip20pt\Delta G < 0 \]
We can use these ideas for find the standard Gibb's Energy change for a chemical change. Free energy is a state function and therefore can be calculated via the free energies of formation of the reactants and products just like the enthalpy of reaction was:
\[\Delta G_{\rm rxn}^\circ =\sum{n\Delta G^\circ_{\rm f} (\rm products)} - \sum{n\Delta G^\circ_{\rm f} (\rm reactants)}\]
Most thermodynamic tables include \(\Delta H^\circ_{\rm f}\), \(\Delta G^\circ_{\rm f}\), and \(\Delta S^\circ\), but sometimes you might NOT have \(\Delta G^\circ_{\rm f}\) (like on an exam) and you should know how to calculate \(\Delta G\) from \(\Delta H\) and \(\Delta S\) using the familiar equation:
\[\Delta G = \Delta H - T\Delta S\]
The standard version looks like this:
\[\Delta G^\circ = \Delta H^\circ - T\Delta S^\circ \]
If something (reaction or process) lowers the free energy, we say such a process is "spontaneous." That simply means it will tend to happen - much like a ball tends to roll downhill. The reverse of this situation is also evident - if a process is "not spontaneous," then it will tend to not happen. This isn't to say that non-spontaneous processes/reactions never happen, it is just that if they are to happen, some external amount of work must be applied to make the process happen - much like physically taking the ball up the hill to a higher position (the ball will never do this without the aid of applied work).
So when we say "it will never happen" for a non-spontaneous process. What we mean is it will "never happen in isolation." If you can combine this process with another one that is spontaneous, then you can make the total or net process spontaneous.
In chemistry, we are often looking at reactions. For these, we compare the pure products and the pure reactants (in their standard state). This comparison gives us the standard change in free energy for a reaction. If this standard free energy change is negative at a given temperature then we would call it "spontaneous." If it is positive, then it is not spontaneous (the reverse reaction is spontaneous). The standard change tells us about the spontaneity of going from all reactants to all products. However, in the real world reactions "end" somewhere in between these two extremes. This is an important concept called chemical equilibrium. Therefore, we say for reactions that are spontaneous (\(-\Delta G\)) "favor the products" side of the equation; those that are not spontaneous (\(+\Delta G\)) "favor the reactants."
Chemical equilibrium is an example of a dynamic equilibrium and not static equilibrium. Know the difference in the two. Static equilibrium is fixed and non-changing – like balancing weights on a balance beam. Dynamic equilibrium has no NET overall change but does have some given processes still proceeding. The process itself proceeds both forwards and backwards at exactly the same rate. Anything that you are constantly depleting via one process is simultaneously being replenished by another process. Stated chemically, equilibrium is achieved when the forward rate of reaction equals the reverse rate of reaction. That is a purely kinetic argument for equilibrium and we will study reaction kinetics (rates) in the second half of general chemistry (CH302). A complete understanding of equilibrium requires knowledge of both arguments (definitions) for the equilibrium state. This unit is about thermodynamics, which has our other definition of equilibrium. It is based purely on thermodynamic state functions. Lets get the thermodynamic argument for equilibrium established next.
The bottom line for the thermodynamic argument lies in the spontaneity of a reaction. The Second Law of Thermodynamics dictates what direction of change is the spontaneous direction. We know that indicator to be universal entropy (\(\Delta S_{\rm univ}\)), although in the previous section we showed how the free energy change (\(\Delta G\)) is an even better indicator for us because it is a purely system based state function. Once again, lets look at the definition of \(G\).
\[G = H - T\;S\]
You'll notice that \(G\) is made up from 3 other state functions. Also pay close attention to how \(G\) will change with \(T\). LOOK at the equation, as \(T\) increases, \(G\) must decrease. If we hold temperature (\(T\)) and pressure (\(P\)) constant, we get the following for the change in free energy:
\[\Delta G = \Delta H - T\Delta S\]
This powerful formula allows us to track spontaneity with a purely system state function, \(\Delta G\). It tracks via sign the opposite of the way that \(\Delta S_{\rm univ}\) does because \(\Delta G = -T\Delta S_{\rm univ}\). Now consider the 3 possible mathematical outcomes for \(\Delta G\):
\[\Delta G < 0\]
⊖ negative
spontaneous
\[\Delta G = 0\]
zero
equilibrium
\[\Delta G > 0\]
⊕ positive
non-spontaneous
We now have a new standard to judge spontaneity and equilibrium. ALL equilibrium processes must have a free energy change equal to zero. This is the same as saying that all the free energies (that’s plain ol’ \(G\) here) of all the reactants must equal the free energies of all the products – our “stalemate” condition for equilibrium.
Notice how \(\Delta G\)’s sign is controlled by the signs on \(\Delta H\) and \(\Delta S\). There are four distinct cases here. The following plot shows each of these cases:
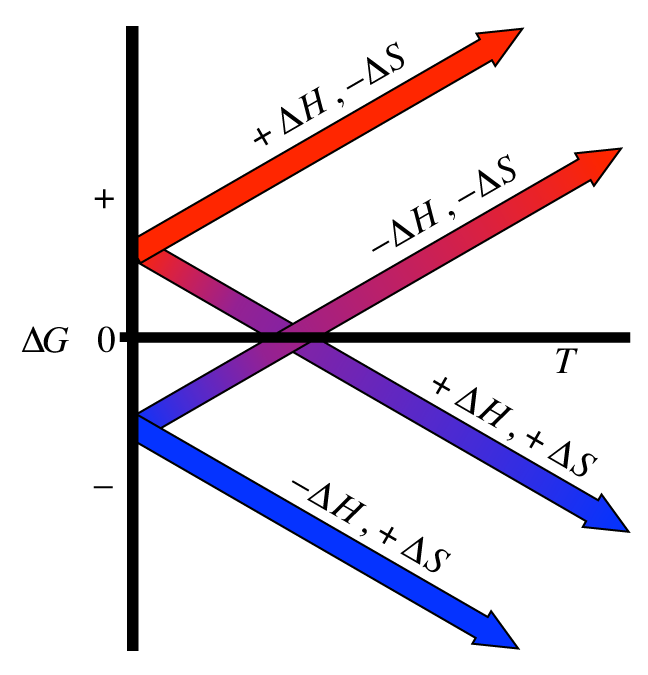
Note there are only 2 of those cases where equilibrium is even possible; those are the cases where the 2 plots cross the zero-point for \(\Delta G\). This is only when \(\Delta H\) and \(\Delta S\) have matching signs. Remember that we typically control the temperature of the reactions that we are running. Note how by controlling temperature, we, in effect, control the spontaneity as well.
Phase transitions occurring at their standard temperature (ice melting at 0 °C are reversible equilibrium processes. If you have an equilibrium process occurring, then \(\Delta G = 0\) and therefore at these temperatures
\(\displaystyle{\Delta H = T\Delta S}\) \(\displaystyle{\Delta S = {\Delta H \over T}}\) \(\displaystyle{T = {\Delta H \over \Delta S }}\)
For example, for water \(\Delta S_{fusion} = (\Delta H_{fusion} / 273.15 K)\) since the temperature for the phase transition is 0 °C (273.15 K). You can also use this idea to find the phase transition temperature if you know the enthalpy and the entropy changes.